Before doing real calculations, several parameters (e.g. energy cut-off, k-points, lattice, …) must be checked to ensure the quality of calculations. This article provides sciripts about convergence test based on ASE (version >= 3.14.0) and VASP 5.4.1. Different styles will be adopted to exemplify different ase-vasp features.
ENCUT test
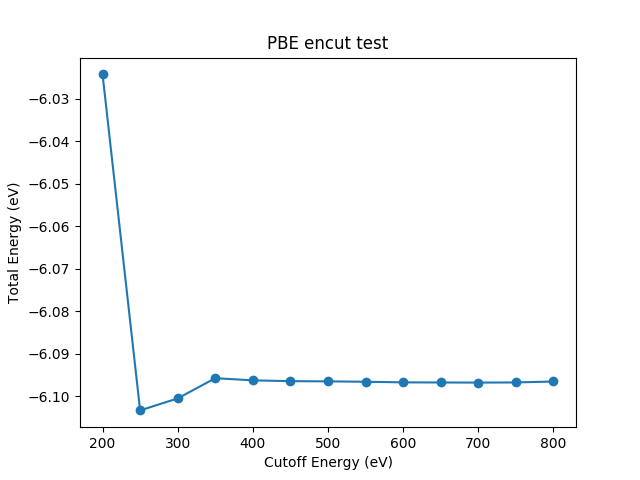
ENCUT (energy cut-off) test requires a series of total energy calculations on various ENCUT values
|
|
If there is a problem with generating figures (_tkinter.TclError: couldn't connect to display "localhost:11.0"),
the following code should be added before matplotlib is used
For ENCUT test, the choice of k-points does not affect the convergence trend of ENCUT. A moderate k-points would be fine. When ENCUT reaches 400 eV, the energy difference between this point and previous point is less than 1 meV/atom. Then 400 eV energy cut-off can be choosen for further calculation. This criterion depends on your demand. Generally, I like to create different directories for different calculations, since label feature is not currently supported in ase-vasp calculator. Someone is working on VASP with FileIOCalculator. But it takes time to make changes, because the ase-vasp calculator is used by a large number of people and small changes can affect many users.
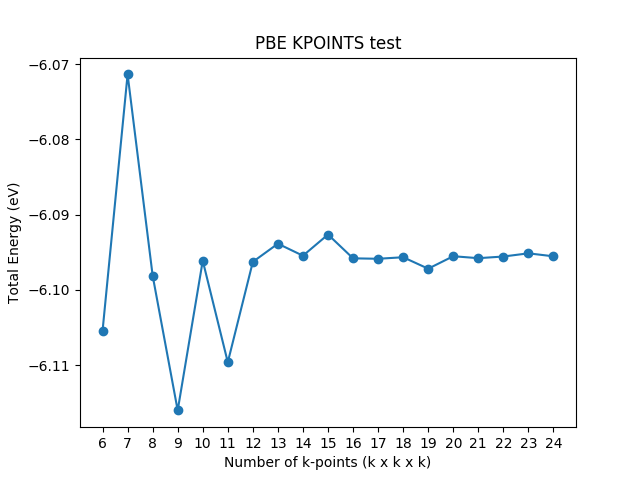
k-points test
The strategy for k-points test is the same as ENCUT test and the difference lies in loop variable.
Lattice test
Four types of lattice test are given here.
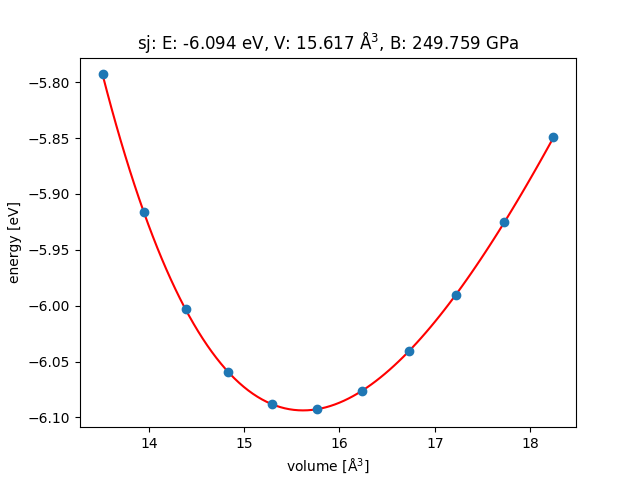
Pt
Lattice test of FCC metal (e.g. Pt) is easy to perform with only one variable needed to be considered.
First, you need to perform a series of total energy calculations at different lattice constants.
Then, fit equation of state.
Finally, optimised lattice constant can be found.
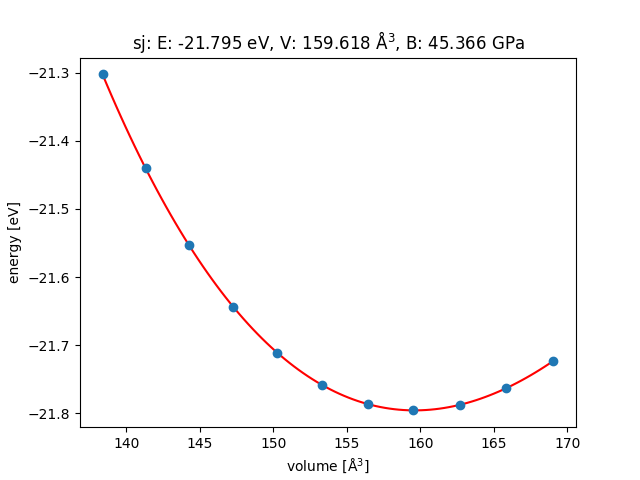
MoS2
Structure optimization has to be done to get energy miminum, otherwise errors would be large.
|
|
TiO2
It’s convenient to check structure with a calculate flag. traj file is used for data analysis.
The chance of making mistakes will be reduced if caculation part and analysis part are seperated.
ZrO2
For system containing more than 2 variables, it’s suggested to do a full-relaxtion (optimising both cell and atoms) with high accuracy